
Detection of QTLs Regulating Six Agronomic Traits of Rice Based on Chromosome Segment Substitution Lines of Common Wild Rice (Oryza rufipogon Griff.) and Mapping of qPH1.1 and qLMC6.1

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Development of the DP15-CSSLs
2.3. The Methods of Phenotypic Characterization of DP15-CSSLs
2.4. Whole Genome Re-Sequencing (WGRS) and Bioinformatic Analysis of Genomic SSR, InDel, and SNP Markers
2.5. Genomic DNA Extraction and PCR Amplification
2.6. QTL Mapping and Data Analysis
3. Results
3.1. Whole Genome Re-Sequencing (WGRS) of the Parental Materials
3.2. Selection of Polymorphic Markers between DP15 and 93-11 Genome
3.3. Chromosome Substitution Segments Analysis of DP15-CSSLs
3.4. Characteristics of Four Grain Related Traits of the DP15-CSSLs
3.5. Identification and Detection of TGW-Related QTLs of DP15-CSSLs
3.6. Identification and Detection of GL-Related QTLs of DP15-CSSLs
3.7. Identification and Detection of GW-Related QTLs of DP15-CSSLs
3.8. Identification and Detection of GLWR-Related QTLs of DP15-CSSLs
3.9. Identification and Genetic Mapping of the qPH1.1
3.9.1. Characterization of the PH of a DP15-CSSL Line
3.9.2. Characterization of the Cell Morphology in Culm
3.9.3. Genetic Analysis and Mapping of the qPH1.1
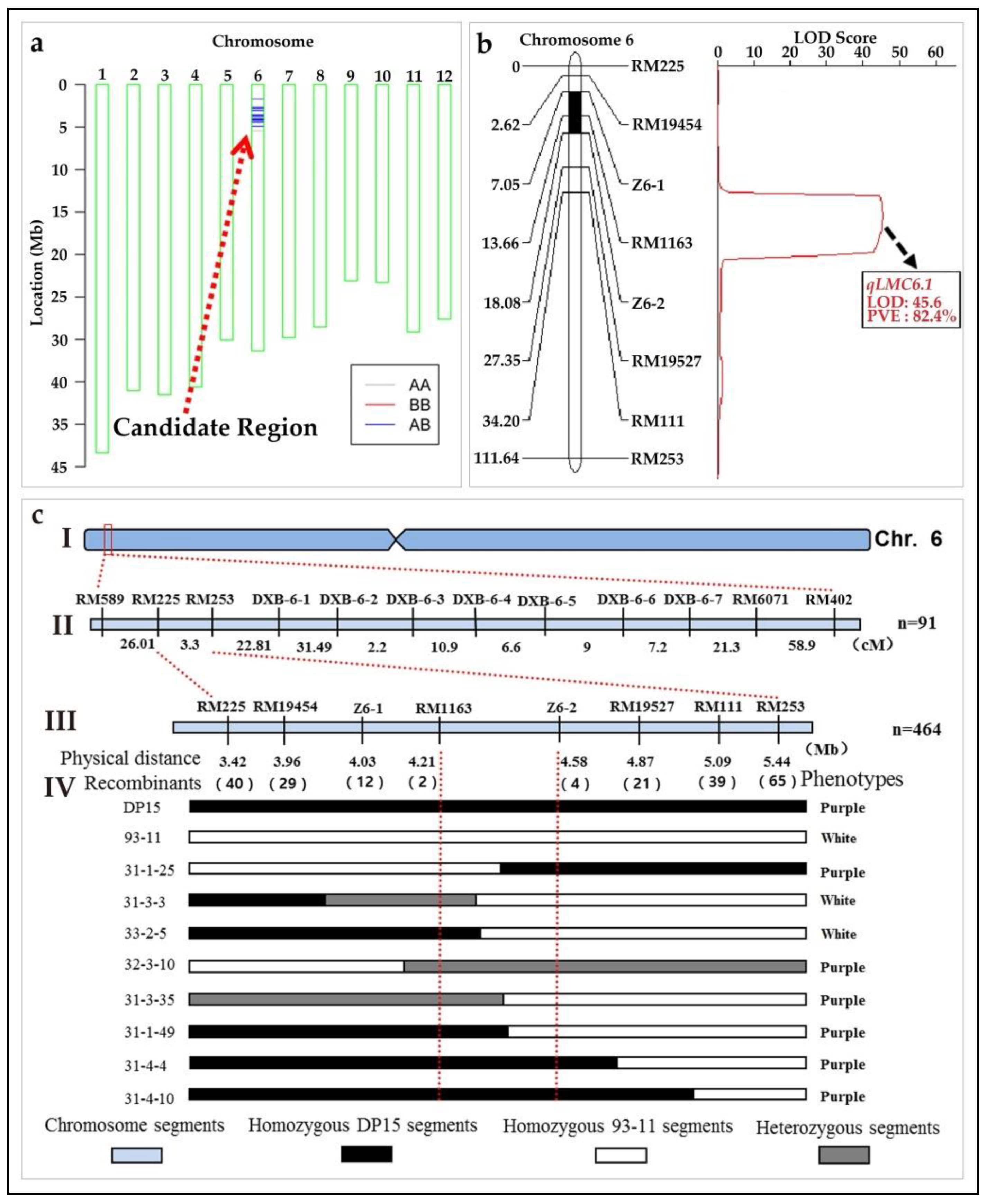
3.10. Identification and Genetic Mapping of the qLMC6.1
3.10.1. Characterization of the LMC of a DP15 CSSLs Line
3.10.2. Characterization of the Cell Morphology in Stigma Cell
3.10.3. Genetic Analysis and Mapping of the qLMC6.1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CTAB | Hexadecyl Trimethyl Ammonium Bromide |
| PH | Plant Height |
| GW | Grain Width |
| GL | Grain Length |
| GN | Grain Number |
| TGW | Thousand-grain Weight |
| GLWR | Grain Length-width Ratio |
| LMC | Leaf Margin Color |
| AC | Apiculus Color |
| SC | Stigma Color |
| LSC | Leaf Sheath Color |
| SSR | Simple Sequence Repeats |
| SNP | Single Nucleotide Polymorphism |
| InDel | Insertion-Deletion |
| RFLP | Restriction Fragment Length Polymorphism |
| MAS | Marker-assisted Selection |
| PAGE | Polyacrylamide Gel Electrophoresis |
| PCR | Polymerase Chain Reaction |
| CSSLs | Chromosome Segment Substitution Lines |
| SSSLs | Single Segment Substitution Lines |
| NILs | Near Iso-genic Lines |
| DHs | Double Haploid Lines |
| LOD | Logarithm of The Odds |
| PVE | Phenotypic Variation Explained |
| QTL | Quantitative Trait Locus |
| BSA | Bulked Segregants Analysis Sequencing |
| Chr. | Chromosome |
| RNA-seq | RNA-Sequencing |
| WGRS | Whole Genome Re-sequencing |
| GWAS | Genome Wide Association Study |
| BSA | Bulked Segregants Analysis |
| GA | Gibberellin Acid |
| SEM | Scanning Electron Microscope |
| MAPK | Mitogen-activated protein kinase |
| bHLH | Basic Helix-loop-helix Transcription Factor |
| UTR | Untranslated Regions |
| CDS | Coding DNA sequence |
| ncRNA | Non-coding RNA |
| CAD | Cinnamyl Alcohol Dehydrogenase |
| Mt | Mitochondrion |
| Pt | Chloroplast |
References
- Zhang, J.; Long, Y.; Xue, M.; Xiao, X.; Pei, X. Identification of microRNAs in Response to Drought in Common Wild Rice (Oryza rufipogon Griff.) Shoots and Roots. PLoS ONE 2017, 12, e0170330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, D.T.; Buu, B.C.; Lang, N.T.; Toriyama, K.; Nakamura, I.; Ishikawa, R. Genetic diversity among perennial wild rice Oryza rufipogon Griff., in the Mekong Delta. Ecol. Evol. 2019, 9, 2964–2977. [Google Scholar] [CrossRef] [Green Version]
- Baucom, R.S.; Estill, J.C.; Leebens-Mack, J.; Bennetzen, J.L. Natural selection on gene function drives the evolution of LTR retrotransposon families in the rice genome. Genome Res. 2009, 19, 243–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigueira, C.C.; Qi, X.; Song, B.K.; Li, L.F.; Caicedo, A.L.; Jia, Y.; Olsen, K.M. Call of the wild rice: Oryza rufipogon shapes weedy rice evolution in Southeast Asia. Evol. Appl. 2019, 12, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Descalsota-Empleo, G.I.; Noraziyah, A.A.S.; Navea, I.P.; Chung, C.; Dwiyanti, M.S.; Labios, R.J.D.; Ikmal, A.M.; Juanillas, V.M.; Inabangan-Asilo, M.A.; Amparado, A.; et al. Genetic Dissection of Grain Nutritional Traits and Leaf Blight Resistance in Rice. Genes 2019, 10, 30. [Google Scholar] [CrossRef] [Green Version]
- Gouda, G.; Gupta, M.K.; Donde, R.; Mohapatra, T.; Vadde, R.; Behera, L. Marker-assisted selection for grain number and yield-related traits of rice (Oryza sativa L.). Physiol. Mol. Biol. Plants 2020, 26, 885–898. [Google Scholar] [CrossRef]
- Usman, B.; Nawaz, G.; Zhao, N.; Liao, S.; Qin, B.; Liu, F.; Liu, Y.; Li, R. Programmed Editing of Rice (Oryza sativa L.) OsSPL16 Gene Using CRISPR/Cas9 Improves Grain Yield by Modulating the Expression of Pyruvate Enzymes and Cell Cycle Proteins. Int. J. Mol. Sci. 2021, 22, 249. [Google Scholar] [CrossRef]
- Wang, S.; Wu, K.; Yuan, Q.; Liu, X.; Liu, Z.; Lin, X.; Zeng, R.; Zhu, H.; Dong, G.; Qian, Q.; et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet. 2012, 44, 950–954. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, Y.; Mao, F.; Gu, Y.; Tang, Z.; Xin, Y.; Liu, F.; Tang, T.; Gao, H.; Zhao, X. Fine-Tuning of the Grain Size by Alternative Splicing of GS3 in Rice. Rice 2022, 15, 4. [Google Scholar] [CrossRef]
- Tomita, M.; Yazawa, S.; Uenishi, Y. Identification of Rice Large Grain Gene GW2 by Whole-Genome Sequencing of a Large Grain-Isogenic Line Integrated with Japonica Native Gene and Its Linkage Relationship with the Co-integrated Semidwarf Gene d60 on Chromosome 2. Int. J. Mol. Sci. 2019, 20, 5442. [Google Scholar] [CrossRef]
- Qin, M.; Zhang, Y.; Yang, Y.; Miao, C.; Liu, S. Seed-Specific Overexpression of SPL12 and IPA1 Improves Seed Dormancy and Grain Size in Rice. Front. Plant Sci. 2020, 11, 532771. [Google Scholar] [CrossRef] [PubMed]
- Harushima, Y.; Yano, M.; Shomura, A.; Sato, M.; Shimano, T.; Kuboki, Y.; Yamamoto, T.; Lin, S.Y.; Antonio, B.; Parco, A.; et al. A high-density rice genetic linkage map with 2275 markers using a single F2 population. Genetics 1998, 148, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Wu, Y.; Li, W.; Qin, X.; Wang, Y.; Yue, B. Genetic mapping with testcrossing associations and F2:3 populations reveals the importance of heterosis in chilling tolerance at maize seedling stage. Sci. Rep. 2017, 7, 14336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanyange, L.; Kamau, J.; Ombori, O.; Ndayiragije, A.; Muthini, M. Genotyping for Blast (Pyricularia oryzae) Resistance Genes in F2 Population of Supa Aromatic Rice (Oryza sativa L.). Int. J. Genom. 2019, 2019, 5246820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, R.; Zhao, N.; Usman, B.; Luo, L.; Liao, S.; Qin, Y.; Nawaz, G.; Li, R. Development of Chromosome Segment Substitution Lines (CSSLs) Derived from Guangxi Wild Rice (Oryza rufipogon Griff.) under Rice (Oryza sativa L.) Background and the Identification of QTLs for Plant Architecture, Agronomic Traits and Cold Tolerance. Genes 2020, 11, 980. [Google Scholar] [CrossRef]
- Yang, W.J.; Nadolska-Orczyk, A.; Wood, K.V.; Hahn, D.T.; Rich, P.J.; Wood, A.J.; Saneoka, H.; Premachandra, G.S.; Bonham, C.C.; Rhodes, J.C.; et al. Near-isogenic lines of maize differing for glycinebetaine. Plant Physiol. 1995, 107, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Liu, X.; Wang, M.; Meyer, R.S.; Luo, X.; Ndjiondjop, M.; Tan, L.; Zhang, J.; Wu, J.; Cai, H.; et al. A single-nucleotide polymorphism causes smaller grain size and loss of seed shattering during African rice domestication. Nat. Plants 2017, 3, 17064. [Google Scholar] [CrossRef]
- Chen, K.; Łyskowski, A.; Jaremko, Ł.; Jaremko, M. Genetic and Molecular Factors Determining Grain Weight in Rice. Front. Plant Sci. 2021, 12, 605799. [Google Scholar] [CrossRef]
- Xu, F.; Sun, X.; Chen, Y.; Huang, Y.; Tong, C.; Bao, J. Rapid Identification of Major QTLs Associated with Rice Grain Weight and Their Utilization. PLoS ONE 2015, 10, e0122206. [Google Scholar] [CrossRef] [Green Version]
- Niu, Y.; Chen, T.; Wang, C.; Chen, K.; Shen, C.; Chen, H.; Zhu, S.; Wu, Z.; Zheng, T.; Zhang, F.; et al. Identification and allele mining of new candidate genes underlying rice grain weight and grain shape by genome-wide association study. BMC Genom. 2021, 22, 602. [Google Scholar] [CrossRef]
- Lyu, J.; Wang, D.; Duan, P.; Liu, Y.; Huang, K.; Zeng, D.; Zhang, L.; Dong, G.; Li, Y.; Xu, R.; et al. Control of Grain Size and Weight by the GSK2-LARGE1/OML4 Pathway in Rice. Plant Cell 2020, 32, 1905–1918. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Shi, J.; Ji, Z.; Wen, Z.; Liang, Y.; Yang, C. Combination of twelve alleles at six quantitative trait loci determines grain weight in rice. PLoS ONE 2017, 12, e0181588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Dai, Z.; Li, L.; Wang, J.; Miao, X.; Shi, Z. OsRAMOSA2 Shapes Panicle Architecture through Regulating Pedicel Length. Front. Plant Sci. 2017, 8, 1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Zhao, X.; Dai, Z.; Ma, F.; Miao, X.; Shi, Z. OsmiR396 growth regulating factor modulate rice grain size through direct regulation of embryo-specific miR408. Plant Physiol. 2021, 186, 519–533. [Google Scholar] [CrossRef]
- Zhu, B.; Si, L.; Wang, Z.; Zhu, Y.Z.J.; Shangguan, Y.; Lu, D.; Fan, D.; Li, C.; Lin, H.; Qian, Q.; et al. Genetic Control of a Transition from Black to Straw-White Seed Hull in Rice Domestication. Plant Physiol. 2011, 155, 1301–1311. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.; Qian, Q.; Tang, D.; Wang, K.; Li, M.; Cheng, Z. A mutation in the rice chalcone isomerase gene causes the golden hull and internode 1 phenotype. Planta 2012, 236, 141–151. [Google Scholar] [CrossRef]
- Zhang, K.; Qian, Q.; Huang, Z.; Wang, Y.; Li, M.; Hong, L.; Zeng, D.; Gu, M.; Chu, C.; Cheng, Z. GOLD HULL AND INTERNODE2 Encodes a Primarily Multifunctional Cinnamyl-Alcohol Dehydrogenase in Rice. Plant Physiol. 2006, 140, 972–983. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhang, Z.; Chen, C.; Wu, W.; Ren, N.; Jiang, C.; Yu, J.; Zhao, Y.; Zheng, X.; Yang, Q.; et al. The C–S–A gene system regulates hull pigmentation and reveals evolution of anthocyanin biosynthesis pathway in rice. J. Exp. Bot. 2018, 69, 1485–1498. [Google Scholar] [CrossRef] [Green Version]
- Shoeva, O.; Gordeeva, E.; Khlestkina, E. The Regulation of Anthocyanin Synthesis in the Wheat Pericarp. Molecules 2014, 19, 20266–20279. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Yang, L.; Huang, W.; Luo, X.; Xie, J.; Hu, B.; Chen, Y. Flavonoid Metabolic Profiles and Gene Mapping of Rice (Oryza sativa L.) Purple Gradient Grain Hulls. Rice 2022, 15, 43. [Google Scholar] [CrossRef]
- Ahmadikhah, A.; Marufinia, A. Effect of reduced plant height on drought tolerance in rice. 3 Biotech 2016, 6, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spielmeyer, W.; Ellis, M.H.; Chandler, P.M. Semidwarf (sd-1), “Green Revolution” Rice, Contains a Defective Gibberellin 20-Oxidase Gene. Proc. Natl. Acad. Sci. USA 2002, 99, 9043–9048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Zhang, K.; Huang, M.; Hector, S.B.; Liu, B.; Tong, C.; Liu, Q.; Zeng, J.; Gao, Y.; Xu, T.; et al. Involvement of Fenton chemistry in rice straw degradation by the lignocellulolytic bacterium Pantoea ananatis Sd-1. Biotechnol. Biofuels 2016, 9, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.J.; Kim, J.H.; Lee, H.J.; Kim, D.H.; Yu, J.; Bae, S.; Cho, Y.; Kang, K.K. Generation and Transcriptome Profiling of Slr1-d7 and Slr1-d8 Mutant Lines with a New Semi-Dominant Dwarf Allele of SLR1 Using the CRISPR/Cas9 System in Rice. Int. J. Mol. Sci. 2020, 21, 5492. [Google Scholar] [CrossRef] [PubMed]
- Yamamuro, C.; Ihara, Y.; Wu, X.; Noguchi, T.; Fujioka, S.; Takatsuto, S.; Ashikari, M.; Kitano, H.; Matsuoka, M. Loss of function of a rice brassinosteroid insensitive1 homolog prevents internode elongation and bending of the lamina joint. Plant Cell 2000, 12, 1591–1605. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, M.; Kiyota, S.; Hanada, A.; Yamaguchi, S.; Takano, M. The Multiple Contributions of Phytochromes to the Control of Internode Elongation in Rice. Plant Physiol. 2011, 157, 1187–1195. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.P.; Piskurewicz, U.; Turečková, V.; Carat, S.; Chappuis, R.; Strnad, M.; Fankhauser, C.; Lopez-Molina, L. Spatially and genetically distinct control of seed germination by phytochromes A and B. Genes Dev. 2012, 26, 1984–1996. [Google Scholar] [CrossRef] [Green Version]
- Fang, K.; Yi, X.; Dai, W.; Gao, H.; Cao, L. Effects of Integrated Rice-Frog Farming on Paddy Field Greenhouse Gas Emissions. Int. J. Environ. Res. Public Health 2019, 16, 1930. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Q.; Xu, Y.; Jeppesen, E.; Gu, X.; Mao, Z.; Chen, H. Farming practices affect the amino acid profiles of the aquaculture Chinese mitten crab. PeerJ 2021, 9, e11605. [Google Scholar] [CrossRef]
- Long, P.; Huang, H.; Liao, X.; Fu, Z.; Zheng, H.; Chen, A.; Chen, C. Mechanism and capacities of reducing ecological cost through rice–duck cultivation. J. Sci. Food Agric. 2013, 93, 2881–2891. [Google Scholar] [CrossRef]
- Matsubara, K.; Yonemaru, J.; Kobayashi, N.; Ishii, T.; Yamamoto, E.; Mizobuchi, R.; Tsunematsu, H.; Yamamoto, T.; Kato, H.; Yano, M. A follow-up study for biomass yield QTLs in rice. PLoS ONE 2018, 13, e0206054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsubara, K.; Yamamoto, E.; Kobayashi, N.; Ishii, T.; Tanaka, J.; Tsunematsu, H.; Yoshinaga, S.; Matsumura, O.; Yonemaru, J.; Mizobuchi, R.; et al. Improvement of Rice Biomass Yield through QTL-Based Selection. PLoS ONE 2016, 11, e0151830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercati, F.; Sunseri, F. Genetic Diversity Assessment and Marker-Assisted Selection in Crops. Genes 2020, 11, 1481. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Cheng, Y.; Han, S.; Van Handel, B.; Dong, L.; Li, X.; Xie, X. Whole genome sequencing and comparative transcriptome analysis of a novel seawater adapted, salt-resistant rice cultivar-sea rice 86. BMC Genom. 2017, 18, 147. [Google Scholar] [CrossRef] [Green Version]
- Basava, R.K.; Hash, C.T.; Mahendrakar, M.D.; Kishor, P.B.K.; Satyavathi, C.T.; Kumar, S.; Singh, R.B.; Yadav, R.S.; Gupta, R.; Srivastava, R.K. Discerning combining ability loci for divergent environments using chromosome segment substitution lines (CSSLs) in pearl millet. PLoS ONE 2019, 14, e0218916. [Google Scholar] [CrossRef] [Green Version]
- Beyene, Y.; Gowda, M.; Olsen, M.; Robbins, K.R.; Pérez-Rodríguez, P.; Alvarado, G.; Dreher, K.; Gao, S.Y.; Mugo, S.; Prasanna, B.M.; et al. Empirical Comparison of Tropical Maize Hybrids Selected Through Genomic and Phenotypic Selections. Front. Plant Sci. 2019, 10, 1502. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Mackon, E.; Jeazet Dongho Epse Mackon, G.C.; Zhao, Y.; Li, Q.; Dai, X.; Yao, Y.; Xia, X.; Nong, B.; Liu, P. Combined Analysis of BSA-Seq Based Mapping, RNA-Seq, and Metabolomic Unraveled Candidate Genes Associated with Panicle Grain Number in Rice (Oryza sativa L.). Biomolecules 2022, 12, 918. [Google Scholar] [CrossRef]
- Kersey, Y.M.; Wessells, N.K. Localization of actin filaments in internodal cells of characean algae. A scanning and transmission electron microscope study. J. Cell Biol. 1976, 68, 264–275. [Google Scholar] [CrossRef] [Green Version]
- Ellison, M.A.; McMahon, M.B.; Bonde, M.R.; Palmer, C.L.; Luster, D.G. In situ hybridization for the detection of rust fungi in paraffin embedded plant tissue sections. Plant Methods 2016, 12, 37. [Google Scholar] [CrossRef] [Green Version]
- Mackon, E.; Ma, Y.; Jeazet Dongho Epse Mackon, G.C.; Li, Q.; Zhou, Q.; Liu, P. Subcellular Localization and Vesicular Structures of Anthocyanin Pigmentation by Fluorescence Imaging of Black Rice (Oryza sativa L.) Stigma Protoplast. Plants 2021, 10, 685. [Google Scholar] [CrossRef]
- Gadin, J.R.; van’T Hooft, F.M.; Eriksson, P.; Folkersen, L. AllelicImbalance: An R/ bioconductor package for detecting, managing, and visualizing allele expression imbalance data from RNA sequencing. BMC Bioinform. 2015, 16, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backman, T.W.H.; Girke, T. SystemPipeR: NGS workflow and report generation environment. BMC Bioinform. 2016, 17, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordon-Thaden, I.E.; Chanderbali, A.S.; Gitzendanner, M.A.; Soltis, D.E. Modified CTAB and TRIzol protocols improve RNA extraction from chemically complex Embryophyta. Appl. Plant Sci. 2015, 3, 1400105. [Google Scholar] [CrossRef] [PubMed]
- Brunel, D. An alternative, rapid method of plant DNA extraction for PCR analyses. Nucleic Acids Res. 1992, 20, 4676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevallet, M.; Luche, S.; Rabilloud, T. Silver staining of proteins in polyacrylamide gels. Nat. Protoc. 2006, 1, 1852–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, Y.; Chung, C.; Lee, Y.; Na, H.; Lee, J.; Lee, S.; Lee, K.; Yoon, Y.; Kang, J.; Lee, H.; et al. QTL Mapping of Grain Quality Traits Using Introgression Lines Carrying Oryza rufipogon Chromosome Segments in Japonica Rice. Rice 2016, 9, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Lei, L.; Li, P.; Wang, J.; Wang, C.; Yang, F.; Chen, J.; Liu, H.; Zheng, H.; Xin, W.; et al. Identification of Candidate Genes Conferring Cold Tolerance to Rice (Oryza sativa L.) at the Bud-Bursting Stage Using Bulk Segregant Analysis Sequencing and Linkage Mapping. Front. Plant Sci. 2021, 12, 647239. [Google Scholar] [CrossRef]
- McCouch, S.R. Gene Nomenclature System for Rice. Rice 2008, 1, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Jaari, S.; Li, M.H.; Merila, J. A first-generation microsatellite-based genetic linkage map of the Siberian jay (Perisoreus infaustus): Insights into avian genome evolution. BMC Genom. 2009, 10, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Wang, X.; Mo, Y.; Li, Y.; Yan, L.; Li, Z.; Shu, W.; Cheng, L.; Huang, F.; Qiu, Y. Genetic analysis and fine mapping of the gall midge resistance gene Gm5 in rice (Oryza sativa L.). Theor. Appl. Genet. 2020, 133, 2021–2033. [Google Scholar] [CrossRef]
- Liu, J.M.; Park, S.J.; Huang, J.; Lee, E.J.; Xuan, Y.H.; Je, B.I.; Kumar, V.; Priatama, R.A.; Raj K, V.; Kim, S.H.; et al. Loose Plant Architecture1 (LPA1) determines lamina joint bending by suppressing auxin signalling that interacts with C-22-hydroxylated and 6-deoxo brassinosteroids in rice. J. Exp. Bot. 2016, 67, 1883–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, G.; Park, J.; Yoon, J.; Yu, S.; An, G. A RING finger E3 ligase gene, Oryza sativa Delayed Seed Germination 1 (OsDSG1), controls seed germination and stress responses in rice. Plant Mol. Biol. 2010, 74, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Ma, Y.; Li, J. The rice YABBY4 gene regulates plant growth and development through modulating the gibberellin pathway. J. Exp. Bot. 2016, 67, 5545–5556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, X.; Wu, S.; Zhu, Z.; Liu, F.; Fu, Y.; Cai, H.; Sun, X.; Gu, P.; Xie, D.; Tan, L.; et al. NOG1 increases grain production in rice. Nat. Commun. 2017, 8, 1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagomi, K.; Shigemune, A.; Sasahara, H.; Arai, T.; Hirabayashi, H.; Yamanouchi, U.; Ideta, O. Finding a novel QTL responsible for kernel cracking resistance from CSSLs of ‘Itadaki’ (O. sativa L.) × donor O. rufipogon. Breed. Sci. 2020, 70, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Surapaneni, M.; Balakrishnan, D.; Mesapogu, S.; Addanki, K.R.; Yadavalli, V.R.; Tripura Venkata, V.G.N.; Neelamraju, S. Identification of Major Effect QTLs for Agronomic Traits and CSSLs in Rice from Swarna/Oryza nivara Derived Backcross Inbred Lines. Front. Plant Sci. 2017, 8, 1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Zhou, K.; Xiang, S.; Zhang, Q.; Li, R.; Li, M.; Liang, P.; Farkhanda, N.; He, G.; Ling, Y.; et al. Identification, pyramid and candidate genes of QTLs for associated traits based on a dense erect panicle rice CSSL-Z749 and five SSSLs, three DSSLs and one TSSL. Rice 2021, 14, 55. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, T.; Wang, H.; Zhao, N.; Liu, B. Heritable alteration in salt-tolerance in rice induced by introgression from wild rice (Zizania latifolia). Rice 2012, 5, 36. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Su, J.; Xu, M.; Zhou, Z.; Zhu, X.; Ma, X.; Hou, J.; Tan, L.; Zhu, Z.; Cai, H.; et al. A common wild rice-derived BOC1 allele reduces callus browning in indica rice transformation. Nat. Commun. 2020, 11, 443. [Google Scholar] [CrossRef] [Green Version]
- Cen, W.; Zhao, W.; Ma, M.; Lu, S.; Liu, J.; Cao, Y.; Zeng, Z.; Wei, H.; Wang, S.; Li, R.; et al. The Wild Rice Locus CTS-12 Mediates ABA-Dependent Stomatal Opening Modulation to Limit Water Loss Under Severe Chilling Stress. Front. Plant Sci. 2020, 11, 575699. [Google Scholar] [CrossRef]
- Cen, W.; Liu, J.; Lu, S.; Jia, P.; Yu, K.; Han, Y.; Li, R.; Luo, J. Comparative proteomic analysis of QTL CTS-12 derived from wild rice (Oryza rufipogon Griff.), in the regulation of cold acclimation and de-acclimation of rice (Oryza sativa L.) in response to severe chilling stress. BMC Plant Biol. 2018, 18, 163. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Chen, L.; Zhao, Y.; Guo, H.; Li, J.; Rashid, M.A.R.; Lu, C.; Zhou, W.; Yang, X.; Liang, Y.; et al. Natural Variation in OsMKK3 Contributes to Grain Size and Chalkiness in Rice. Front. Plant Sci. 2021, 12, 784037. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Yamamoto, T.; Shimizu, T.; Ma, X.F.; Shomura, A.; Takeuchi, Y.; Lin, S.Y.; Yano, M. Genetic dissection and pyramiding of quantitative traits for panicle architecture by using chromosomal segment substitution lines in rice. Theor. Appl. Genet. 2008, 116, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Qiao, W.; Qi, L.; Cheng, Z.; Su, L.; Li, J.; Sun, Y.; Ren, J.; Zheng, X.; Yang, Q. Development and characterization of chromosome segment substitution lines derived from Oryza rufipogon in the genetic background of O. sativa spp. indica cultivar 9311. BMC Genom. 2016, 17, 580. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Han, B.; Tang, J.; Zhang, J.; Cui, D.; Geng, L.; Zhou, H.; Li, M.; Han, L. Construction of chromosome segment substitution lines of Dongxiang common wild rice (Oryza rufipogon Griff.) in the background of the japonica rice cultivar Nipponbare (Oryza sativa L.). Plant Physiol. Biochem. 2019, 144, 274–282. [Google Scholar] [CrossRef]
- Bessho-Uehara, K.; Furuta, T.; Masuda, K.; Yamada, S.; Angeles-Shim, R.B.; Ashikari, M.; Takashi, T. Construction of rice chromosome segment substitution lines harboring Oryza barthii genome and evaluation of yield-related traits. Breed. Sci. 2017, 67, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Furuta, T.; Uehara, K.; Angeles-Shim, R.B.; Shim, J.; Ashikari, M.; Takashi, T. Development and evaluation of chromosome segment substitution lines (CSSLs) carrying chromosome segments derived from Oryza rufipogon in the genetic background of Oryza sativa L. Breed. Sci. 2014, 63, 468–475. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, J.; Wang, X.; Li, R.; Chen, B. Proteomic response of hybrid wild rice to cold stress at the seedling stage. PLoS ONE 2018, 13, e0198675. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, L.; Zhang, Y.; Cao, C.; Liu, F.; Huang, F.; Qiu, Y.; Li, R.; Lou, X. Map-based cloning and characterization of BPH29, a B3 domain-containing recessive gene conferring brown planthopper resistance in rice. J. Exp. Bot. 2015, 66, 6035–6045. [Google Scholar] [CrossRef] [Green Version]
- Du, B.; Zhang, W.; Liu, B.; Hu, J.; Wei, Z.; Shi, Z.; He, R.; Zhu, L.; Chen, R.; Han, B.; et al. Identification and characterization of Bph14, a gene conferring resistance to brown planthopper in rice. Proc. Natl. Acad. Sci. USA 2009, 106, 22163–22168. [Google Scholar] [CrossRef]
- Fujino, K.; Nishimura, T.; Kiuchi, H.; Hirayama, Y.; Sato, T. Phenotypic changes during 100-year rice breeding programs in Hokkaido. Breed. Sci. 2017, 67, 528–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Xue, Y.; Zhou, H.; Li, Y.; Usman, B.; Jiao, X.; Wang, X.; Liu, F.; Qin, B.; Li, R.; et al. High-resolution mapping and breeding application of a novel brown planthopper resistance gene derived from wild rice (Oryza. rufipogon Griff). Rice 2019, 12, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, W.; Wu, H.; Guo, T.; Liu, X.; Wan, X.; Jin, J.; Hanh, T.T.T.; Thoa, N.T.N.; Chen, M.; et al. Fine mapping of stable QTLs related to eating quality in rice (Oryza sativa L.) by CSSLs harboring small target chromosomal segments. Breed. Sci. 2011, 61, 338–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, S.J.S.R.; Singh, K.; Umakanth, B.; Vishalakshi, B.; Rao, K.V.S.; Suneel, B.; Sharma, S.K.; Kadambari, G.K.M.; Prasad, M.S.; Senguttvel, P.; et al. Identification and Characterization of a Large Effect QTL from Oryza glumaepatula Revealed Pi68(t) as Putative Candidate Gene for Rice Blast Resistance. Rice 2020, 13, 17. [Google Scholar] [CrossRef]
- Fuchs, E.J.; Martínez, A.M.; Calvo, A.; Muñoz, M.; Arrieta-Espinoza, G. Genetic diversity in Oryza glumaepatula wild rice populations in Costa Rica and possible gene flow from O. sativa. PeerJ 2016, 4, e1875. [Google Scholar] [CrossRef] [Green Version]
- Cui, D.; Tang, C.; Lu, H.; Li, J.; Ma, X.; A, X.; Han, B.; Yang, Y.; Dong, C.; Zhang, F.; et al. Genetic differentiation and restricted gene flow in rice landraces from Yunnan, China: Effects of isolation-by-distance and isolation-by-environment. Rice 2021, 14, 54. [Google Scholar] [CrossRef]
- Liu, Q.; Qin, J.; Li, T.; Liu, E.; Fan, D.; Edzesi, W.M.; Liu, J.; Jiang, J.; Liu, X.; Xiao, L.; et al. Fine Mapping and Candidate Gene Analysis of qSTL3, a Stigma Length-Conditioning Locus in Rice (Oryza sativa L.). PLoS ONE 2015, 10, e0127938. [Google Scholar] [CrossRef]
- Heang, D.; Sassa, H. Antagonistic actions of HLH/bHLH proteins are involved in grain length and weight in rice. PLoS ONE 2012, 7, e31325. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Sun, Y.; Li, J.; Su, L.; Zheng, X.; Wang, X.; Li, K.; Yang, Q.; Qiao, W. Identify QTLs for grain size and weight in common wild rice using chromosome segment substitution lines across six environments. Breed. Sci. 2017, 67, 472–482. [Google Scholar] [CrossRef] [Green Version]
- Nan, J.; Feng, X.; Wang, C.; Zhang, X.; Wang, R.; Liu, J.; Yuan, Q.; Jiang, G.; Lin, S. Improving rice grain length through updating the GS3 locus of an elite variety Kongyu 131. Rice 2018, 11, 21. [Google Scholar] [CrossRef]
- Fan, C.; Xing, Y.; Mao, H.; Lu, T.; Han, B.; Xu, C.; Li, X.; Zhang, Q. GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor. Appl. Genet. 2006, 112, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiong, G.; Hu, J.; Jiang, L.; Yu, H.; Xu, J.; Fang, Y.; Zeng, L.; Xu, E.; Xu, J.; et al. Copy number variation at the GL7 locus contributes to grain size diversity in rice. Nat. Genet. 2015, 47, 944–948. [Google Scholar] [CrossRef]
- Liu, S.; Hua, L.; Dong, S.; Chen, H.; Zhu, X.; Jiang, J.; Zhang, F.; Li, Y.; Fang, X.; Chen, F. OsMAPK6, a mitogen-activated protein kinase, influences rice grain size and biomass production. Plant J. 2015, 84, 672–681. [Google Scholar] [CrossRef]
- Guo, T.; Chen, K.; Dong, N.; Shi, C.; Ye, W.; Gao, J.; Shan, J.; Lin, H. GRAIN SIZE AND NUMBER1 Negatively Regulates the OsMKKK10-OsMKK4-OsMPK6 Cascade to Coordinate the Trade-off between Grain Number per Panicle and Grain Size in Rice. Plant Cell 2018, 30, 871–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Li, S.; Liu, Q.; Wu, K.; Zhang, J.; Wang, S.; Wang, Y.; Chen, X.; Zhang, Y.; Gao, C.; et al. The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat. Genet. 2015, 47, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Yuan, X.; Wang, Y.; Yang, Y.; Zhang, M.; Yu, H.; Xu, Q.; Wang, S.; Niu, X.; Wei, X. Validation of a QTL for Grain Size and Weight Using an Introgression Line from a Cross between Oryza sativa and Oryza minuta. Rice 2021, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- Xin, F.; Zhu, T.; Wei, S.; Han, Y.; Zhao, Y.; Zhang, D.; Ma, L.; Ding, Q. QTL Mapping of Kernel Traits and Validation of a Major QTL for Kernel Length-Width Ratio Using SNP and Bulked Segregant Analysis in Wheat. Sci. Rep. 2020, 10, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dastidar, K.G.; Maitra, S.; Goswami, L.; Roy, D.; Das, K.P.; Majumder, A.L. An Insight into the Molecular Basis of Salt Tolerance of L-myo-Inositol 1-P Synthase (PcINO1) from Porteresia coarctata (Roxb.) Tateoka, a Halophytic Wild Rice. Plant Physiol. 2006, 140, 1279–1296. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Liu, H.; Zhou, T.; Gu, B.; Huang, X.; Shangguan, Y.; Zhu, J.; Li, Y.; Zhao, Y.; Wang, Y.; et al. An-1 Encodes a Basic Helix-Loop-Helix Protein That Regulates Awn Development, Grain Size, and Grain Number in Rice. Plant Cell 2013, 25, 3360–3376. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Lu, D.; Li, C.; Luo, J.; Zhu, B.; Zhu, J.; Shangguan, Y.; Wang, Z.; Sang, T.; Zhou, B.; et al. Genetic Control of Seed Shattering in Rice by the APETALA2 Transcription Factor SHATTERING ABORTION1. Plant Cell 2012, 24, 1034–1048. [Google Scholar] [CrossRef]
- Su, S.; Hong, J.; Chen, X.; Zhang, C.; Chen, M.; Luo, Z.; Chang, S.; Bai, S.; Liang, W.; Liu, Q.; et al. Gibberellins orchestrate panicle architecture mediated by DELLA-KNOX signalling in rice. Plant Biotechnol. J. 2021, 19, 2304–2318. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, G.; Usman, B.; Zhao, N.; Han, Y.; Li, Z.; Wang, X.; Liu, Y.; Li, R. CRISPR/Cas9 Directed Mutagenesis of OsGA20ox2 in High Yielding Basmati Rice (Oryza sativa L.) Line and Comparative Proteome Profiling of Unveiled Changes Triggered by Mutations. Int. J. Mol. Sci. 2020, 21, 6170. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Liu, Z.; Fu, X.; Dai, Z.; Wang, S.; Zhang, G.; Zeng, R.; Liu, G. Detection and characterization of epistasis between QTLs on plant height in rice using single segment substitution lines. Breed. Sci. 2015, 65, 192–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, A.; Fujioka, S.; Sunohara, H.; Kamiya, N.; Hong, Z.; Inukai, Y.; Miura, K.; Takatsuto, S.; Yoshida, S.; Ueguchi-Tanaka, M.; et al. The Role of OsBRI1 and Its Homologous Genes, OsBRL1 and OsBRL3, in Rice. Plant Physiol. 2006, 140, 580–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morinaka, Y.; Sakamoto, T.; Inukai, Y.; Agetsuma, M.; Kitano, H.; Ashikari, M.; Matsuoka, M. Morphological Alteration Caused by Brassinosteroid Insensitivity Increases the Biomass and Grain Production of Rice. Plant Physiol. 2006, 141, 924–931. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Wu, C.; Yuan, S.; Yin, L.; Sun, W.; Zhao, Q.; Zhao, B.; Li, X. Kinase activity of OsBRI1 is essential for brassinosteroids to regulate rice growth and development. Plant Sci. 2013, 199–200, 113–120. [Google Scholar] [CrossRef]
- Wang, Z.; Cheng, D.; Fan, C.; Zhang, C.; Zhang, C.; Liu, Z. Cell Type-Specific Differentiation Between Indica and Japonica Rice Root Tip Responses to Different Environments Based on Single-Cell RNA Sequencing. Front. Genet. 2021, 12, 659500. [Google Scholar] [CrossRef]
- Helal, G.A. Bioconversion of straw into improved fodder: Mycoprotein production and cellulolytic acivity of rice straw decomposing fungi. Mycobiology 2005, 33, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Silva, S.; Costa, E.M.; Calhau, C.; Morais, R.M.; Pintado, M.E. Anthocyanin extraction from plant tissues: A review. Crit. Rev. Food Sci. Nutr. 2017, 57, 3072–3083. [Google Scholar] [CrossRef]
- Alappat, B.; Alappat, J. Anthocyanin Pigments: Beyond Aesthetics. Molecules 2020, 25, 5500. [Google Scholar] [CrossRef]
- Mackon, E.; Jeazet Dongho Epse Mackon, G.C.; Ma, Y.; Kashif, M.H.; Ali, N.; Usman, B.; Liu, P. Recent Insights into Anthocyanin Pigmentation, Synthesis, Trafficking, and Regulatory Mechanisms in Rice (Oryza sativa L.) Caryopsis. Biomolecules 2021, 11, 394. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Lee, Y.; Byeon, E.; Kang, B.; Kyeoung, D.; Kim, C. Whole-genome resequencing and transcriptomic analysis of genes regulating anthocyanin biosynthesis in black rice plants. 3 Biotech 2018, 8, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.H.; Park, S.; Lee, J.Y.; Ha, S.H.; Lee, J.G.; Lim, S.H. A Rice B-Box Protein, OsBBX14, Finely Regulates Anthocyanin Biosynthesis in Rice. Int. J. Mol. Sci. 2018, 19, 2190. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, N.; Yuan, R.; Usman, B.; Qin, J.; Yang, J.; Peng, L.; Mackon, E.; Liu, F.; Qin, B.; Li, R. Detection of QTLs Regulating Six Agronomic Traits of Rice Based on Chromosome Segment Substitution Lines of Common Wild Rice (Oryza rufipogon Griff.) and Mapping of qPH1.1 and qLMC6.1. Biomolecules 2022, 12, 1850. https://doi.org/10.3390/biom12121850
Zhao N, Yuan R, Usman B, Qin J, Yang J, Peng L, Mackon E, Liu F, Qin B, Li R. Detection of QTLs Regulating Six Agronomic Traits of Rice Based on Chromosome Segment Substitution Lines of Common Wild Rice (Oryza rufipogon Griff.) and Mapping of qPH1.1 and qLMC6.1. Biomolecules. 2022; 12(12):1850. https://doi.org/10.3390/biom12121850
Chicago/Turabian StyleZhao, Neng, Ruizhi Yuan, Babar Usman, Jiaming Qin, Jinlian Yang, Liyun Peng, Enerand Mackon, Fang Liu, Baoxiang Qin, and Rongbai Li. 2022. "Detection of QTLs Regulating Six Agronomic Traits of Rice Based on Chromosome Segment Substitution Lines of Common Wild Rice (Oryza rufipogon Griff.) and Mapping of qPH1.1 and qLMC6.1" Biomolecules 12, no. 12: 1850. https://doi.org/10.3390/biom12121850